
MBNL Sequestration Links Myotonic Dystrophy to Autism via RNA Mis-Splicing
Expanded CUG Repeats in DMPK Disrupt Neural Splicing of Autism-Risk Genes During Brain Development

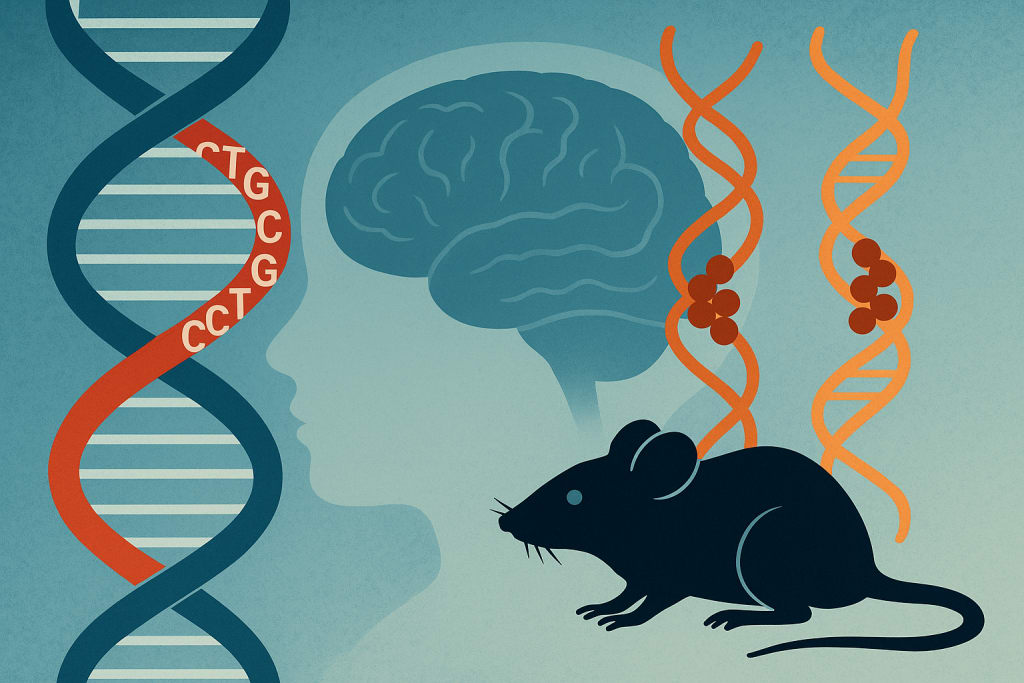
Abstract
Autism spectrum disorder has been linked to enrichment of gene-specific tandem repeat expansions across the genome. The CTG tandem repeat expansion in the DMPK gene's 3′ untranslated region, which is known to cause myotonic muscular dystrophy type 1, is one such mutation. Myotonic dystrophy and autism have a clear clinical connection, but the molecular basis for this connection is unknown. During brain development, the RNA splicing patterns of autism-risk genes, particularly a class of autism-relevant microexons, are altered when mutant DMPK RNAs with expanded CUG repeats sequester MBNL splicing factors. We show that both DMPK-CTG expansion and Mbnl null mouse models replicate autism-relevant mis-splicing profiles, as well as altered responses to novelty and social behavioral deficits. Our hypothesis that developmental mis-splicing of autism-risk genes causes myotonic dystrophy-associated autism is supported by these findings.
Main
Autism spectrum disorder (ASD) is a neurodevelopmental condition that affects communication, social interactions, limited interests, and repetitive behaviors1. It is genetically and clinically diverse1. Over 95% of children with ASD have at least one additional physical or mental health condition2,3. ASD affects one in every 36 children. The molecular mechanisms that explain ASD and comorbid conditions remain elusive, despite the fact that hundreds of genes are known to increase risk for ASD4. Tandem repeat mutations that increase the risk of ASD5,6 were found in recent whole-genome sequencing studies. A CTG expansion (CTGexp) in the 3′ untranslated region of the DM1 protein kinase (DMPK) gene is one of the most frequent mutations. A CTG expansion (CTGexp) is defined as having more than 50 CTG repeats. This mutation is thought to affect about 1 in 2,100 newborns7. Myotonic dystrophy type 1 (DM1) is a neuromuscular disease caused by the DMPK-CTGexp mutation. Symptom severity varies greatly and can begin as early as infancy8. A molecular explanation for the manifestation of ASD in DM1-affected individuals is unknown; however, previous studies reported comorbidity of DM1 and ASD and demonstrated that the presence of ASD is associated with younger age of DM1 onset9,10,11,12,13. Numerous high-affinity binding sites for muscleblind-like (MBNL) RNA-binding proteins (RBPs) are provided by DMPK-CUGexp transcripts in DM1, resulting in the sequestration of MBNL and the formation of biomolecular condensates known as RNA foci14,15. During embryonic stem cell pluripotency and reprogramming, cell type differentiation and maturation, and organ development16,17,18, MBNL proteins, including MBNL1 and MBNL2, are trans-acting factors that regulate alternative splicing (AS). A wide range of DM1 clinical symptoms are brought on by MBNL loss in postnatal tissues, which results in a "adult-to-fetal" reversion of the AS program19,20. The CUGexp length, the concordant sequestration level of MBNL paralogs, and the degree of mis-splicing all well match the severity of DM1 symptoms21,22. DM1 and ASD exhibit developmental missplicing23,24,25. Mis-splicing of ASD-risk genes has been linked to RBFOX and SRRM4 RBPs, according to previous research26,27. SRRM4 mostly controls the inclusion of neuronal microexons (miEs) of 3 to 30 nucleotides (nt), which are misregulated in 30% of idiopathic ASD brains26, whereas RBFOX controls multiple AS types. By encoding sites for post-translational modification and regulating protein–protein interaction networks, MiEs are crucial to the development of the nervous system28,29. Mice with ASD-like behavioral phenotypes, including social avoidance30,31, can be affected by modifications that recapitulate neuronal miE mis-splicing. The mechanistic pathway that underlies ASD traits is still unknown, despite the fact that numerous DM1 mouse models have been studied32,33. Through the CUGexp repeat in DM1, we present a mechanistic understanding of autism here.
We show that splicing disruptions are caused by MBNL protein loss, with 17–25% of these occurring in known ASD-risk genes, including multiple top ASD-risk genes like SCN2A and ANK2. In DM1, we discover that neuronal miEs belong to a new category of mis-spliced events. MiE mis-splicing of ASD-risk genes caused by SRRM4 protein downregulation in idiopathic ASD is replicated by sequestration of MBNL proteins. The synergistic regulation of the ANK2 miE by the MBNL and SRRM proteins is demonstrated by our in-depth mechanistic analysis. Finally, we used both Dmpk-CTGexp knock-in (KI) and Mbnl2 knock-out (KO) mouse models to demonstrate impairments in social behavior. According to our findings, the loss of CUGexp-induced MBNL activity results in developmental mis-splicing of ASD-linked genes in DM1-associated ASD.
WRITTED BY ME
About the Creator
Mahafuj Alam
🗞️ Mahafuj Alam | News Curator & Independent Media Voice
delivering news quickly, accurately, and bravely, including headline breaking stories and untold local stories. Stay informed. Keep your power.
Keep reading
More stories from Mahafuj Alam and writers in Humans and other communities.
Theory of Cell-by-Cell Color Perception in Oz Vision
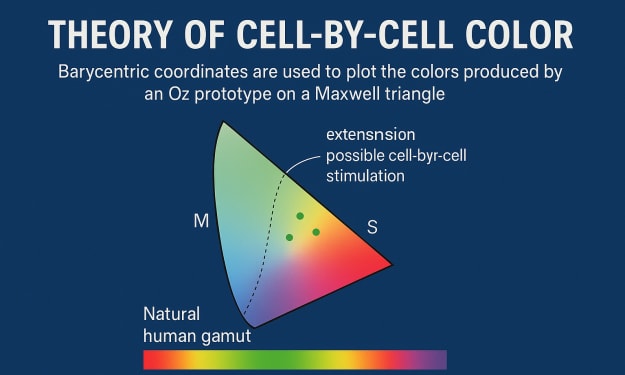
Theory of color from cell to cell Barycentric coordinates are used to plot the colors produced by our Oz prototype on a Maxwell triangle (19). . This triangle projects its total activation (L, M, and S) while simultaneously displaying the color's chromaticity in two dimensions (2D). The natural human gamut, which encompasses all chromaticities that are attainable through normal spectral mixtures of light, is depicted in these diagrams as a color-filled subregion plotted at the bottom. In theory, the fundamentally larger color gamut that is accessible via cell-by-cell stimulation in Oz is the full area of the chromaticity triangle, assuming ideal conditions that produce perfect localization of light to target cones. However, in practice, a small amount of light will miss target cones and stimulate nearby cells, shifting the activation pattern away from the intended Oz color and toward the laser's natural color. In Fig., it is predicted how this stray cone activation will affect the colors that can be achieved. 2Opens in a picture viewer The point-spread function (PSF) of the laser microdoses on the retina in relation to the spacing of cone cells, the cone's spatial light-gathering function (20–22), errors in microdose targeting during eye movement, the proportion of L:M:S cones in the retina, and the stimulating wavelength are the most important factors. In the image viewer, Figure 2AOpens shows how the range of Oz chromaticities that are possible would be affected by fractional light leak. When stimulating the retina at 4° eccentricity, as shown, a diffraction-limited PSF would theoretically enable Oz to address nearly all possible chromaticities in the lms triangle, but not in the foveola, where cone cells are at their smallest. In reality, the total leakage of light includes more than just diffraction because of things like residual aberrations following adaptive optics focusing and microdose targeting errors caused by computational latency during eye movement. It is difficult to measure these factors directly, but the model shown in Fig. 2AOpens compared to the experimental color matching data in the upcoming "Color matching experiments" section suggest that two-thirds of the light captured by cones is captured by neighboring cones, while one-third is confined to the target cell. In spite of this unintentional light leak, our Oz prototype displays color beyond the normal human gamut with this level of accuracy. In the image viewer, Figure 2BOpens shows how the range of Oz chromaticities that could be achieved would be affected by the stimulating wavelength. Depending on the number of different cone types that respond to a given stimulating wavelength, the shape of this gamut can be a triangle, a line, or a single point. This shape reflects the relative responses of the L, M, and S cone cells. Prototype design An AOSLO (14), which simultaneously images and stimulates the retina using a raster scan of near-diffraction-limited laser light over a 0.9° square field of view, serves as the foundation for our Oz prototype. We are able to monitor the eye's movement in real time by imaging the retina with almost invisible infrared light. We dynamically target each cone cell in the field of view with pulses of visible-wavelength laser light to compensate for this motion. A population of 103 cones receives these laser microdoses at a rate of 105 per second. The spectral type of each cone needs to be known in order for cone-targeted stimulation to result in the intended LMS activation. Using recently developed optoretinography techniques in an AO-OCT system, cone cells in the subject's retina are categorized according to their spectral type in a preparatory step (15, 23). We use a classified region with 1000 to 2000 cones near a 4° eccentricity from the foveola for this study. We conduct the following experiments on human subjects: color matching with uniform Oz color squares and image/video recognition. The stimulated area falls within the classified region of the retina because all Oz stimuli are presented within the 0.9° square field of view, 4° adjacent to a gaze fixation point. Stimuli are randomly repeated in a control condition, and the microdose delivery is intentionally compromised. Each microdose in these control trials is randomly "jittered" so that it lands two cones away from the target. Studies on matching colors In order to officially measure the chromaticity coordinates of Oz colors, we carry out color matching experiments. Both 488 nm, which can activate all three L, M, and S cone types, and 543 nm, which is close to the peak of L and M but only minimally activates S, are tested as stimulation wavelengths. A red-green-blue (RGB) projector and a near-monochromatic laser with a tunable wavelength that can be mixed with white projector light are the two color matching systems we use. The latter is able to produce colors that are close to the natural human gamut, making it clear that our attempts to display olo do not actually go beyond the natural human gamut. The subject must judge match equality using the same patch of retina in order to eliminate effects from differences in adaptation across the retina because the subject sees 0.9° squares of Oz and controllable color that coincide in space and alternate in time during a color matching trial. Subjects can, as is typical with color matching (1), add light to the Oz color—also known as "negative" light—if necessary to achieve an exact match; the matched color is calculated by subtracting its color coordinates from those of the controllable square. In addition, subjects are asked to qualitatively name the hue and rate the saturation (on a scale of one to four) of the squares of controllable color and Oz color. The color matching experiments' graphs are shown in Figure 3 (opens in an image viewer). 222 color matches were performed by five subjects. We draw attention to four observations. First, around the stimulation wavelength of 488 nm, Oz colors form a triangle (Fig. 3BOpens in the image viewer), as well as a 543 nm color line (Fig. 3AOpens in the image viewer), which is in line with the theory in the section titled "Theory of cell-by-cell color." Second, as anticipated, the color "collapses" toward the stimulation wavelength under the jitter control condition. Thirdly, the further Oz colors are from the color matching system's gamut, the greater the variance in matching lms chromaticity. This pattern is in line with the geometric analysis in the Materials and Methods section titled "Plotting perceptual uncertainty in matching," which explains why perceptual uncertainty in chromaticity rises when light must be added to the test color to match. Fourth, Picture The image viewer's 3COpens provide unambiguous evidence that olo goes beyond the normal human spectrum. To match the (nearly) monochromatic colors shown, which are on the boundary of the natural human gamut, all subjects found it necessary to desaturate olo with projector white in these matches. Under the test subjects' viewing conditions, these matching monochromatic wavelengths between 501 and 512 nm are the most saturated teal hues for normal color vision. Although the Abney effect—a shift in hue with saturation (24)—opens the possibility that the hue of the wavelength at best match may not precisely represent the hue of the undiluted olo color, subjects' qualitative hue naming and saturation ratings support these quantitative findings. The color names "teal," "green," "blue-greenish," and "green, a little blue" were all suggested for olo. Fig.'s near-monochromatic colors have an average saturation rating of 2.9, while subjects consistently give olo a saturation rating of 4. 3Creates in the image viewer. Experiments in image and video recognition In order to investigate the capacity of human subjects to comprehend Oz-generated images, we create image and video recognition experiments. We use four-alternative forced choice (4-AFC) and two-alternative forced choice (AFC) tasks in which participants can only succeed if they use accurate Oz stimulation-generated hue information. Subjects must determine the image's line orientation in the 4-AFC task. Subjects must identify the direction of rotation in a video of a moving disk in the 2-AFC task. With a stimulating wavelength of 543 nm, these stimuli present the lines and disks as red (all-L cone) on an olo background (all-M cone). In the jitter control condition, all hue and luminance cues are eliminated and the task is reduced to guessing after a calibration step is carried out (see the "Image and video recognition experiments" section in Materials and Methods). The results of the 4-AFC line orientation and 2-AFC rotation direction tasks are plotted in Figure 4Opens in an image viewer. Subjects are able to accurately detect both line orientation and motion direction (blue bars) in the experimental condition. Subjects' performance in the jitter control condition is reduced to guessing for both tasks (gray bars). When the task was easy, subjects report seeing red or orange lines and disks on a blue-green or green background, whereas when they were forced to guess, they see a yellow-green square. The first is directly related to precise Oz microdose delivery, and the second is related to the jitter control condition, in which only the natural color of the 543-nm light should be seen. DISCUSSION
By Mahafuj Alam10 months ago in Humans
Time to change your life.
Is it even possible for you to change your life? A lot of people, more than there should be, are unhappy with their lives. But what are they doing about it? Usually nothing. People talk about how shitty their lives are and want change, but are they not taking the actions to change?
By Jen Phillipsa day ago in Humans
How to Get Over Emotional Cheating Without Losing Yourself
Emotional cheating cuts deep. It may not necessarily require physical contact, but it frequently hurts just as much. Trust crumbles softly. Doubt settles very quickly. Many people feel lost, confused, and unsure of who they are following the event. On the Bloom Boldly platform, we believe that you can heal without shrinking or losing your voice. We may move on with confidence, clarity, and self-respect.
By Bloom Boldly4 days ago in Humans



Comments (1)
Actually this sickness of ASD is very sad, I haven friend of mine Son, how their suffering to recover Son. Good article, you have written fantastically.